Press Release: DLRC Group Becomes a Certified B Corporation
Published Dec 02, 2025
Published 30th January 2024
In the ever-evolving landscape of the withdrawal of the United Kingdom (UK) from the European Union (EU), staying abreast of the latest frameworks and requirements is crucial for businesses to thrive. One such framework that is to be implemented on 1 January 2025 is the Windsor Framework.
Following the exit of the UK from the EU in 2020, the Northern Ireland Protocol (NIP) established systems for trade in and out of Northern Ireland (NI) mostly through the continued application of EU law, but has led to a mass of practical difficulties both in the flow of goods across the Irish Sea to NI, and legal compliance for businesses operating in the province. The new framework offers solutions that, if implemented, promise to address many of these problems.
Although a single medicines regulatory framework for the entire UK is welcomed by many, companies may yet again have to adjust their supply chains, in particular for Centrally Authorised Products (CAPs) that currently have a common/joint pack for NI and other EU Member States.
In this article, we will delve into the updated licensing, labelling, and packaging requirements associated with the Windsor Framework and explore how businesses can navigate these changes to ensure compliance.
For more than two years NI has faced difficulties trading with the wider UK. The Northern Ireland Protocol was an integral part of the withdrawal agreement when the UK exited from the EU and established a single regulatory zone on the island of Ireland, by applying specific EU legislation “to and in” NI, thereby preventing a hard border between NI and Ireland.
In terms of medicines licensing, this has meant that products currently placed on the market in NI need to comply with EU pharmaceutical rules rather than those of the UK, and medicines entering NI from GB are viewed as an import from a third country. Products destined for NI therefore require either a UK-wide Marketing Authorisation or NI Marketing Authorisation issued by MHRA in compliance with EU rules, or an EU centralised Marketing Authorisation granted by the European Commission (EC) following review by the European Medicines Agency (EMA). The application of UK medicines rules in GB, and EU rules in NI, has led to variations in review timelines and product information, with the potential for inequality in patient access, alongside continued disruption, and uncertainty for medicinal product supply chains into NI from GB.
In recognition of the difficulties incurred across many sectors due to the Northern Ireland Protocol, on 27 February 2023 the UK government and the European Commission reached an agreement, the Windsor Framework, designed to simplify trade between GB and NI while safeguarding the EU single market. For medicines, this is achieved by making it harder for GB- and NI-authorised products to be supplied in EU countries. Additional detail on the impact of the Windsor Framework on medicines licensing has been released by MHRA over the course of 2023, and more recently by EMA. Further updates, particularly in relation to pharmacovigilance activities, are expected in early 2024. The new arrangements suggest a fundamental change to the existing system under the Northern Ireland Protocol.
A key objective of the Windsor Framework is to ensure a permanent solution to safeguarding the supply and access of medicines from GB into NI.
The Windsor Framework will be implemented from 1 January 2025. In order to better understand the operational impacts, key requirements related to licensing, and to labelling and packaging are presented below.
Implementation of the Windsor Framework will allow the MHRA to license all medicines, including medicines that were historically under the mandatory scope of the EU Centralised Authorisation Procedure (CAP), across the whole of the UK under one UK-wide regime. All new and innovative medicines, orphan medicinal products, Advanced Therapy Medicinal Products (ATMP) and paediatric medicines authorised will be granted UK product licences (PLs). Most new Marketing Authorisations will be granted as “PL” licences covering the whole of the UK and the Marketing Authorisation number will have a PL prefix (Table 1).
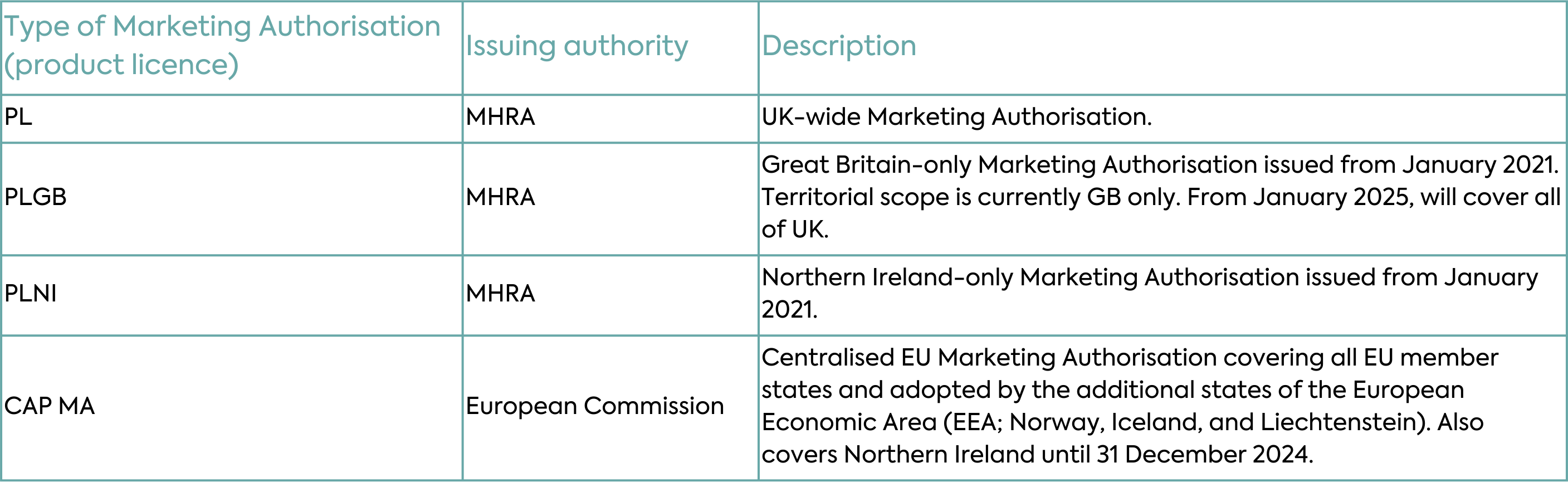
With respect to existing Marketing Authorisations, the changes under the Windsor Framework will depend on the type of authorisation. Typical scenarios for new and existing Marketing Authorisations are depicted in Table 2 and described in detail below.
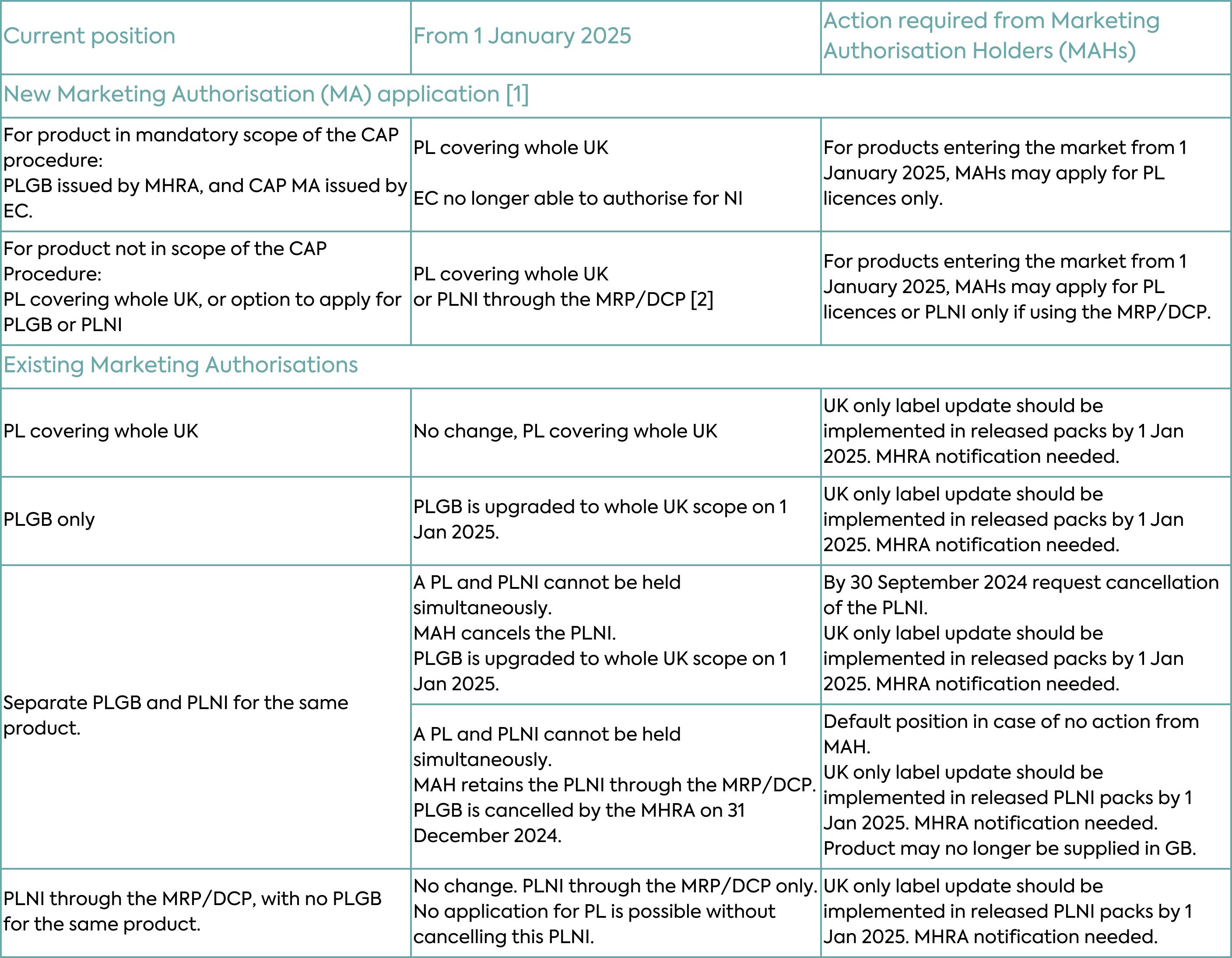
PLGB Marketing Authorisations (products licensed for GB only and granted by the MHRA since the UK left the EU) will automatically become valid for the whole of the UK on 1 January 2025, as MHRA takes on responsibility for these medicines in Northern Ireland. The change in territorial scope will not require a change in licence number, and these medicines will retain the PLGB prefix. PLGBs will become something of a historical oddity, a piece of “Brexit” history. No action will be required from Marketing Authorisation Holders for this conversion, however, Marketing Authorisation Holders need to comply with the associated labelling and packaging requirements as discussed in more detail below.
With the expansion in scope of PLGBs, Marketing Authorisation Holders could theoretically hold a PLGB and PLNI (a product licensed by the MHRA that covers NI only as the territorial application) both of which would cover NI. This is not permissible, so if a Marketing Authorisation Holder currently holds both a PLNI and a PLGB for the same product and wishes to retain UK-wide authorisation, the PLNI will need to be cancelled. Only then can the existing PLGB be converted to a UK-wide Marketing Authorisation. Importantly, the MHRA deadline to receive a request to cancel the PLNI is 30 September 2024. If a cancellation request is not received, the MHRA will automatically cancel the PLGB. This means that only the PLNI will remain from 1 January 2025 i.e. the medicine may no longer be supplied to GB after that date.
PLNI Marketing Authorisations (products licensed for NI only and granted by the MHRA) will not be automatically expanded in scope in the same way. Where a Marketing Authorisation Holder only holds a PLNI and subsequently seeks authorisation for the whole of the UK, the PLNI must be cancelled prior to the granting of the new PL as a UK-wide Marketing Authorisation. Marketing Authorisation Holders involved in EU procedures, with NI included as a Concerned Member State (CMS), will need to notify the coordinating Reference Member State (RMS) to withdraw NI from the procedure. This must occur before the cancellation of the PLNI and the granting of the new PL.
Existing UK-wide Marketing Authorisation granted by MHRA through EU collaborative assessment procedures (Mutual Recognition Procedure or Decentralised Procedure) prior to the UK leaving the EU, and where NI remains involved as a Concerned Member State, are not affected by the Windsor Framework. They can continue to be managed in alignment with the EU procedure, unless there is a requirement for UK-specific changes, which would then require withdrawal of NI from the procedure and granting of a UK-wide Marketing Authorisation.
Finally, MHRA anticipates that the CAP Bridging Mechanism and Northern Ireland MHRA Authorised Route (NIMAR) schemes, implemented to mitigate potential gaps in medicines availability in NI, will no longer be required to support the supply of medicines to NI. The CAP Bridging Mechanism will therefore end after 31 December 2024. The Northern Ireland MHRA Authorised Route (NIMAR) will remain in place in law (but not used).
A fundamental principle of the Windsor Framework is that there is a clear distinction between medicines destined for the UK internal market compared to those entering the EU single market. To preclude onward movement of medicines licensed on a UK-wide basis into any part of the EU, while ensuring all medicines use the same packaging and labelling across the UK, all medicines placed on the UK market from implementation of the Windsor Framework on 1 January 2025 must:
This means a switch in sourcing of medicines. From 1 January 2025, the NI market can no longer be supplied with EU-authorised products and must be supplied under a UK product licence. EU/EEA packs (labelled for the EU and/or European Economic Area markets) in existing packaging and already Qualified Person (QP)-released into the NI supply chain as of 1 January 2025, can continue to be marketed until their expiry without any relabelling or repackaging. However, these medicinal products cannot be moved from NI to an EU/EEA Member State or be placed on the market in an EU/EEA Member State.
No further Qualified Person (QP) release of EU/EEA packs to NI, or NI packs to EU/EEA may occur after 1 January 2025.
The latest MHRA Guidance (updated 11 January 2024) and EMA Questions and Answers to Stakeholders (issued 5 January 2024) provide further detail on the above measures.
The MHRA Guidance specifies the following with respect to the “UK only” label:
The EU Falsified Medicines Directive (FMD) places an obligation on manufacturers of prescription-only medicinal products supplied in EU/EEA (and currently also NI) to apply certain safety features to reduce the risk of falsified (“fake”) medicines entering the supply chain. The EU Falsified Medicines Directive does not apply in GB and will cease to apply in NI from 1 January 2025. This means that serialisation codes, included on a medicine’s outer packaging and registered with the European Medicines Verification Organisation (EMVO) for compliance with the EU Falsified Medicines Directive, will not be permitted in GB or NI. Marketing Authorisation Holders must therefore ensure these codes are removed or covered in any new UK packs from January 2025.
Marketing Authorisation Holders can however, use similar coding that may be embedded in 2D barcodes (including expiry date, batch number, Global Trade Item Number) and serial numbers that have not been uploaded to the European Medicines Verification Organisation repository. The MHRA continues to encourage the use of other safety features such as anti-tamper devices.
The changes to product labelling, packaging and potentially also to supply chain configuration are significant and will take time to implement. Companies may wish to get ahead.
Marketing Authorisation Holders can begin making the labelling changes required under the Windsor Framework for UK-wide and GB licensed products and notifying the MHRA (how to do this is described below). However, while the packs of UK-wide licensed products (i.e. those to be marketed in both GB and NI) can be updated to display the “UK only” label before 1 January 2025, they must also remain compliant with the requirements of the EU Falsified Medicines Directive until 31 December 2024 as it remains in force for NI until then.
Medicines with GB-only product licences can also be updated early and released to the market in GB before 1 January 2025. Qualified Person-released PLGB packs meeting these requirements will be valid for supply to the Northern Ireland market immediately after 1 January 2025.
Of particular note to companies marketing products throughout the EU and UK is the confirmation that joint EU/UK medicines packs can no longer enter the supply chain from 1 January 2025, as the changes resulting from the Windsor Framework will make the EU and UK labelling requirements incompatible. However where already released to market, and not repackaged or relabelled, they can continue to be supplied until their expiry date.
Information relevant to EU markets will need to be removed from the outer packaging of “UK only” packs, and likewise, information relevant to NI must be removed from EU packs [3]. Note that the Windsor Framework does not prevent the use of shared (EU/UK) printed inner packaging components such as the leaflet, in cases where the text of the EU and UK Marketing Authorisations remain sufficiently aligned.
For products supplied to meet the needs of very small numbers of patients across the UK and EU (e.g., for ultra-rare conditions), this stipulation under the Windsor Framework for separate UK and EU packs may create a significant issue. If Windsor Framework requirements might lead to supply disruption and/or market withdrawal, Marketing Authorisation Holders should contact the European Medicines Agency (EMA) as well as the UK Department of Health and Social Care (DHSC) and MHRA.
The text and layout of the product labelling is assessed by the EU/EEA and UK regulatory authorities as part of the Marketing Authorisation and will need to be amended to reflect the changes introduced under the Windsor Framework. The main changes are described in the sections below.
Information regarding NI within the list of Marketing Authorisation Holders representatives for the EU common annexes (leaflet text) will become obsolete for EU-only packs (NI will come under UK Product Information for the UK pack).
The EMA must be advised of updates to the EU product information. A request for deletion of the local representative for NI should be requested at the earliest opportunity in any regulatory procedure affecting the Annexes of the Marketing Authorisation (e.g. renewal, variation) submitted after 1 July 2024 (provided that this change is only implemented after 1 January 2025). For products with no regulatory procedure affecting the Annexes expected within 36 months, the Marketing Authorisation Holder should submit an Article 61(3) notification. An update to the Quality Review of Documents (QRD) Product Information template that reflects this change is expected to be published by 29 February 2024.
Before implementing new label and leaflet artwork changes, companies must notify the MHRA of updated artwork by 31 December 2024. There are three methods by which Marketing Authorisation Holders may do so:
MHRA notifications may be submitted in groups of up to 25 Marketing Authorisations
For products that are not currently marketed (the labelling is approved in text format only), there is no need to notify an update within these timelines. The Marketing Authorisation Holder should submit the relevant text and artwork update prior to marketing, in line with current procedures.
To navigate the transition smoothly, DLRC experts can support clients to take proactive measures. This includes engaging early to plan and implement a robust strategy to make the licensing and labelling changes required across the product portfolio in regard to the Windsor Framework. Additionally, the DLRC team stays informed about upcoming additions and changes to Windsor Framework guidance and can provide valuable insights into best practices during this transitional period.
If you are a Marketing Authorisation Holder with an existing UK, GB, or NI Marketing Authorisation (PL, PLGB, or PLNI) and/or a Centralised EU Marketing Authorisation (CAP MA) then the following list of considerations and deadlines may provide a framework to begin your impact analysis:

The MHRA published the latest updates to its guidance around the new labelling arrangements of the Windsor Framework on 11 January 2024. The EMA has also issued a series of stakeholder Q&A providing an insight into EMA expectations for implementation. It is important that companies plan for these changes in order to transition in a compliant manner. For some Marketing Authorisation Holders, the changes will require a great deal of time, commitment and investment in order to comply by 1 January 2025 and as such, preparations should already be underway.
DLRC will continue to follow these developments and provide further updates. In the meantime, if you would like to discuss the Windsor Framework and what it means for your UK and EU medicines, please contact us at hello@dlrcgroup.com or use the link below.


Published Dec 02, 2025

Published Nov 14, 2025

Published Oct 20, 2025

Published Oct 01, 2025

Published Oct 01, 2025

Published Oct 01, 2025

Published Oct 01, 2025
Published Oct 01, 2025

Published Sep 29, 2025