EU HTA and JCA: Redefining Market Access Strategy in Europe
Published Feb 26, 2026
Published 31st July 2024
The presence of active pharmaceuticals in the environment is a growing concern. Environmental risk assessment (ERA) of medicinal products for human use (HMP) ensures that the potential effects of pharmaceuticals on the environment are studied and that adequate precautions are taken where specific risks are identified.
The ERA is based on the use of the HMP and the physico-chemical, ecotoxicological and fate properties (degradation, persistence) of its active substance. This article discusses the legal basis, scope and updated requirements for ERA following the implementation of the revised EMA guidance on 01 September 2024.
The legislative requirement for the conduct of ERAs in the EU has been in place over 15 years. In accordance with Article 8(3) of Directive 2001/83/EC, as amended, the potential environmental risks posed by the use of medicinal products shall be evaluated and, on a case-by-case basis, specific arrangements to limit this risk shall be considered.
The original EU guidance on how to perform an ERA for a HMP was issued by EMA 18 years ago in 2006. A concept paper on the revision of this original guidance was released in 2016; the revised (Rev.1) guidance issued for review and comment in 2018, and the final version comes into effect on 01 September 2024.
Due to the extended review period for the updated EMA guidance, applicants have been aware of the majority of “new” concepts since 2018, and hence the changes have been put into practice by many applicants and regulators ever since.
An ERA will be required for all new Marketing Authorisation Applications (MAAs) for a medicinal product submitted through a centralised, mutual recognition, decentralised or national procedure, regardless of their legal basis, which also means that generics are no longer able to apply for waivers to this requirement.
For type II variations, the ERA dossier should be updated if there is an anticipated increase in the environmental exposure, e.g. a new indication which results in an increase in the extent of the use. Following authorisation of an HMP, appropriate information on the ERA is included in the European Public Assessment Report (EPAR), so that this information is accessible to the public.
Under the current requirements, the outcome of the ERA should not constitute a criterion for refusal of a marketing authorisation. However, under the reform to the EU Pharmaceutical legislation proposed by the European Commission, the mandatory requirement for ERA for all companies introducing HMP into the EU market will be reinforced in order to ensure better protection of the environment. Failure of a pharmaceutical company to accurately present an ERA and risk mitigation measures may result in the denial of marketing authorization for the drug. Furthermore, products already on the market before 2005 will also require an ERA, if not already in place.
An ERA is the process of evaluating the potential risks of a HMP on the environment. The objective is to safeguard aquatic and terrestrial ecosystems, including surface water, groundwater, soil, species vulnerable to secondary poisoning, and microbial processes in sewage treatment plants (STPs).
The outcome of an ERA serves as the basis for implementation of measures to minimise the amount of medicinal product released into the environment. Identification of specific risk-minimisation activities to be taken by the user of the medicine and inclusion of relevant information in the product information to facilitate correct disposal of the HMP by patients and healthcare providers. For example, to ensure that the medicine is disposed of in special containers or returned to a pharmacy.
The ERA is based on the use of the product and the physico-chemical, ecotoxicological, and fate properties of its active substance(s). All pharmacologically active substances in the product need to undergo an ERA.
The original 2006 guidance provided by the EMA described the assessment of potential risks to the environment of a drug substance as a step-wise, phased procedure that could be terminated when sufficient information/data was available to either indicate that the drug substance was unlikely to pose a risk to the environment or to identify and sufficiently characterise the potential risks.
Although the overarching concept of the tiered approach has remained the same, the revised 2024 guideline is more comprehensive and provides more explicit explanation on several topics, providing a more consistent and complete approach for the evaluation of potential environmental risks associated with the use of HMP.
The revised guideline also describes the process for identifying potential hazards associated with the drug substance of a HMP and addresses potential precautionary and risk mitigation measures.
Lastly, the 2024 guideline also includes information on the structure of the ERA Report – to be presented in Module 1.6 of the eCTD dossier of the MAA.
For each ERA, both a risk assessment (exposure calculation) and a hazard assessment are required. The risk assessment reflects the possibility of an effect occurring and is an evaluation of both exposure of organisms in the environment to the active substance, and ecotoxicity. The hazard assessment concerns the identification of intrinsic properties of an active substance that could render it harmful to the environment regardless of the levels of exposure. The hazard assessment is performed independently of the exposure calculation (see Figure 1).
The ERA consists of two parts: Phase I is a mandatory assessment based on environmental exposure and general characteristics of the HMP. Phase II consists of a detailed fate and effects assessment, and definitive persistence, bioaccumulation and toxicity (PBT) evaluation of the HMP.
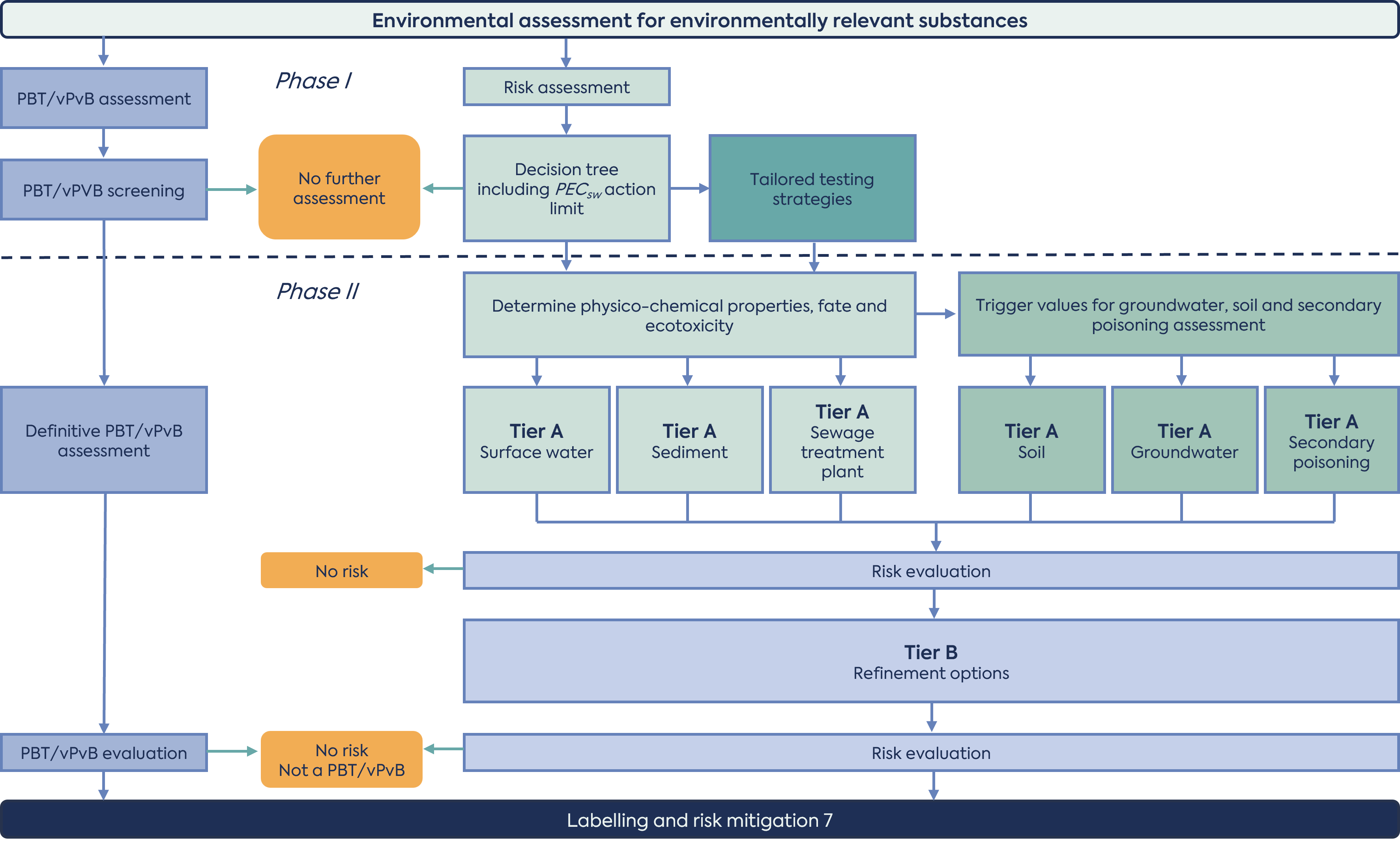
(adapted from the ERA 2024 guideline)
The principal of a tiered approach to ERA is maintained in the revised 2024 guideline, however substantial changes have been incorporated, providing a more consistent and complete approach for the evaluation of potential environmental risks associated with the use of drug substance. Updates have been made to the types/number of test systems recommended and additional test systems/assays are included for areas not, or inadequately, addressed in the 2004 guideline.
Phase I risk assessment is explicitly outlined in a decision tree with a Q&A section, concluding with the calculation of a predicted environmental concentration in water (PECsw). The outcome of phase I is that either the risk assessment stops or that a Phase II assessment is required. The rationale for not proceeding to Phase II should be included in the ERA report. Of note, the decision tree clarifies that a Phase II assessment is not warranted for non-natural peptides/proteins that are readily biodegradable.
The key criteria for entering the Phase II risk assessment are a PECsw of ≥ 0.01 μg/L, with the option to refine the PECsw based on a justification for the fraction of the population receiving the active substance daily (market penetration factor: FPEN). A specific assessment strategy is necessary for some groups of substances, such as antibacterials, antiparasitics and endocrine active substance (EAS). The EAS terminology has been introduced for all compounds that have the potential to affect development or reproduction.
The hazard assessment (PBT/vPvB assessment) concerns the identification of certain intrinsic properties of the active substance, for which the long-term risks to the environment are unpredictable. It identifies active substances that are poorly degraded in the environment (persistent; P), that accumulate in organisms (bioaccumulative; B) and are toxic (T) or very persistent and very bioaccumulative (vPvB). All drug substances (with a few exceptions) must be assessed for PBT properties, regardless of their PEC. A tiered testing strategy should be followed, beginning with a PBT screening assessment (octanol/water partitioning study) in Phase I, with definitive assessment in Phase II if the trigger value is met (logarithmic octanol/water partitioning coefficient (log Kow) >4.5). For substances which do not meet the trigger for definitive PBT/vPvB assessment, an assessment of PBT/vPvB properties may still be required. This will be the case if the outcomes obtained in Phase II of the risk assessment demonstrate that the B- and T-criterion are met, or if the vB-criterion is met.
If a definitive PBT assessment is required, it should follow the PBT and vPvB criteria defined under the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) regulation (Regulation (EC) No 1907/2006).
Phase II risk assessment is where the experimental studies are conducted to allow for a detailed fate and effects assessment. The following studies/assessments should be considered during this phase of the risk assessment:
Phase II assessment consists of two tiers (Figure 1). Tier A requires studies on the physico-chemical properties, environmental fate and ecotoxicological effects of the drug substance. The outcome of these studies determines whether risk assessments for soil, and groundwater and secondary poisoning are necessary. Ultimately, the PEC is then compared to the predicted no effect concentration (PNEC) and, where risks are identified, a Tier B assessment is warranted, which focuses on refining the PEC calculations.
In comparison with the 2006 guideline, most of the fate and effect testing has been moved to Phase II Tier A, with Tier B focusing on refining exposure calculations. A new section on secondary poisoning to estimate the accumulation of drug substances in the food chain has been introduced, and notes that lack of accumulation in mammals does not exclude a potential for accumulation in fish and other aquatic species. Technical details for studies conducted as a part of the Phase II risk assessment are included to ensure consistency in the approach to testing and thus the data provided. In addition, some of the triggers for further assessment and additional studies have been amended.
The principles of the 3Rs (Replacement, Reduction and Refinement) are embedded in the revised 2024 guidance. Drug developers are encouraged to share their data to avoid unnecessary repetition of studies with living organisms. When data is shared a Letter of Access as well as complete reports should be submitted. In the case of missing data, it is the responsibility of the current Applicant to complete the required studies.
Data generated for ERA should be compliant with Good Laboratory Practice (GLP) where applicable and preferably follow the most recent test guidelines issued by the Organisation for Economic Co-operation and Development (OECD) or comparable international validated test guidelines.
A new provision for the use of publicly available data has also been added, which specifies that for drug substances that are already marketed, information may be available in the public domain. A targeted literature review on endpoints of significance is required even if data or a previously performed ERA is obtained from another MAH, to identify new information on ecotoxicity of the active substance. Specific guidance on search, use and evaluation of published data is provided.
The revised EMA Guideline on the ERA of Medicinal Products for human use was issued on the 15 February 2024 and comes into effect on the 01 September 2024.
The revised guideline retains the overarching concept of the tiered approach as per the 2006 guide, however substantial changes have been incorporated to provide a more consistent and thorough approach for the evaluation of potential environmental risks associated with the use of HMP. The guidance also addresses potential precautionary and risk mitigation measures and offers guidance on the structure of the ERA Report.
An ERA will be required for all new MAAs for a HMP regardless of the legal basis of the application. The consequence of this approach to ERA is an equal requirement for all Applicants – and as such there may be a significant impact to generic drug manufacturers where insufficient data are available for older drug substances/generics, and additional testing is required. However, there are now options for better utilisation of data in the public domain to support a scientifically sound assessment of environmental risk, and encouragement to share data in the interest of 3Rs (reduced animal testing), avoiding unnecessary repetition of animal studies (e.g. fish).
DLRC’s team of regulatory experts can advise on current regulations on ERA and author and/or review ERA documents for regulatory submissions. Speak to DLRC today by emailing hello@dlrcgroup.com or use the links below.


Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025

Published Nov 25, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 14, 2025