DLRC Achieves Cyber Essentials Plus Certification
Published Apr 11, 2025
Published 01st August 2024

The FDA’s draft guidance on Diversity Action Plans is open to comments at this stage (comments due by 09/26/2024). Once the final guidance is issued, it will be in effect after 180 days and sponsors will be required to submit Diversity Action Plans with certain clinical trials. Both developers of drugs (including biologics) and devices will be affected. Unless there is an unforeseen delay, these requirements will become applicable for trials enrolling subjects by the end of 2025.
Overall, the guidance is significantly aligned for Diversity Action Plans for drugs and device products but there are differences in process and content. The main focus of Diversity Action Plans is across race, ethnicity, sex, and age, however, the guidance also encourages sponsors to consider other factors such as geographic location, gender identity, sexual orientation, socio-economic status (SES) disabilities, pregnancy and lactation status, and co-morbidity. At this stage, there are no details on actions if enrolment goals are not met. Read on for a summary of the key points from the Guidance for Industry and an explanation of the expected content and process.

Broadly, a Diversity Action Plan is required for studies which will be the primary basis for the FDA review of benefit–risk. More specifically:
Progress updates are expected as part of the Investigational New Drug (IND), or IDE, annual report.
Per the Federal Food, Drug, and Cosmetic Act (FD&C Act), it must include 3 sections, each section is discussed below;
FDA also recommends sponsors share their Diversity Action Plan goals with the public, via the sponsor’s website in consumer-friendly language.
The Diversity Action Plan must include the sponsor’s goals for enrolment in the clinical study, disaggregated by race, ethnicity, sex, and age group of clinically relevant study populations. Goals should be informed by the estimated prevalence or incidence of the condition in the US and recognised sources and methodology should be included to explain the estimates.
The FDA guidance recognises that sponsors make use of multi-regional trials for global development of FDA-regulated products. The FDA recommends that Diversity Action Plans of such programs should describe enrolment goals for the entire study and not only the US-enrolled patients. Furthermore, such programs should be designed to account for the need to enrol a population representative of the US.
To meet the requirement for providing rationale sponsors must include:
The guidance clarifies that the Diversity Action Plan should be focussed on how this particular study will address enrolment and retention strategies to meet diversity goals. The sponsor is encouraged to consult patients and healthcare providers about this.
This should also be accompanied by the sponsor’s plan to monitor enrolment goals during recruitment and conduct.
The Diversity Action Plan is expected to be succinct and to limit cross-referencing to previously submitted documents, overall not exceeding 10 pages.
In rare instances, a sponsor may request the FDA to conduct a case-specific determination regarding the waiving of the requirement to submit a Diversity Action Plan. A full or partial waiver may be granted if defined criteria are met. Such a request can be based on the prevalence or incidence of the condition, conducting the study with a Diversity Action Plan would be impracticable, or a waiver is required to protect public health during a public health emergency. FDA is required to respond within 60 days granting or refusing the waiver request. For drugs, waiver requests should be submitted to the relevant IND in eCTD Module 1.12.5 and for Devices, a stand-alone submission is required.
Diversity Action Plans that were submitted voluntarily to CDER from April 2022 – April 2023 were analysed. As part of Project Equity, the Oncology Division’s analysis of the 91 DAPs submitted found that the majority of plans focussed their efforts on patient-directed measures and community engagement, followed by clinical research workforce-directed measures. This was much more common than changes to eligibility criteria or decentralised trials (Figure 1). Furthermore, of the 41% of Diversity Action Plans for which FDA feedback was sought, 90% of FDA comments were around enrolment goals.
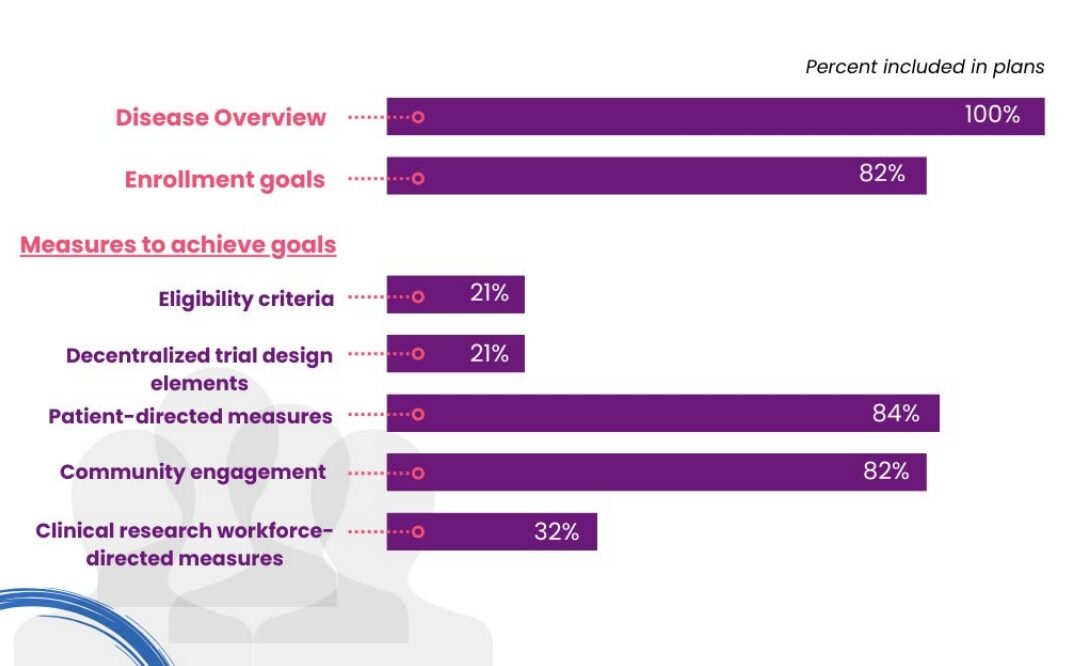
Project Equity – Diversity Plans – Oncology (fda.gov)
Once Diversity Action Plans are a requirement, the FDA will release a report that summarises the Diversity Action Plans received and whether the clinical studies conducted met the demographic enrolment goals. No details are provided on FDA response to Diversity Action Plans that do not meet intended goals.
With this guidance, FDA is setting the expectation for sponsors to engage in cross-program considered planning for enrolling a representative population in clinical studies. Knowledge of the condition and intended target population will drive how goals and measures are shaped. An iterative process is accommodated and should allow Diversity Action Plans to be adapted as product development progresses and a better understanding of the data is acquired. Embedding a new submission into development plans can be a challenge, the FDA advises that feedback on the Diversity Action Plan is most effective if asked alongside questions on design and endpoints. Sponsors of relevant clinical trials planning a future US filing should ensure they are prepared to meet these new requirements and be ready to engage with the FDA. DLRC Inc. have extensive experience interacting with the FDA and can assist with Diversity Action Plans and agency feedback.


Published Apr 11, 2025

Published Mar 27, 2025

Published Mar 25, 2025

Published Mar 06, 2025
Published Feb 26, 2025

Published Feb 25, 2025

Published Feb 03, 2025

Published Feb 03, 2025

Published Jan 30, 2025