Innovating Non-Clinical Testing: NAMs and ESG Impact
Published Jul 21, 2025
Published 24th September 2024

This article provides an overview of the EMA scientific advice and protocol assistance procedure, highlighting the differences between scientific advice and protocol assistance. Protocol assistance is the special form of scientific advice available for developers of designated orphan medicines for rare diseases. The article summarises the key elements of the procedures including pre-submission considerations such as a preparatory meeting, the evaluation phase and post advice actions. We also discuss impact of the EU Pharmaceutical Reform on the scientific advice and protocol assistance procedures.
Scientific advice is a very powerful tool for developers of medicinal products. Not only does it guide the developer through the challenges encountered during development, but it also establishes relationships with regulators. The advice will certainly have a positive impact on the development and will enable the developer to streamline their process, ensure a high rate of compliance with regulations and will reduce the risk of Marketing Authorisation Applications being rejected. Whilst the advice is not legally binding, there will be a high expectation that the advice given by EMA will be addressed during development.
Scientific advice is a process whereby medicine developers can ask EMA specific questions related to their proposed development plan. Questions typically fall into the development strategy, quality, clinical and non-clinical areas although many questions can fall into more than one area, for example qualification of an impurity. The advice is prospective in nature and EMA do not pre-evaluate results which will be submitted later in development.
Protocol assistance is the term for scientific advice sought for medicines with an orphan drug designation. The same principles and process relating to regular scientific advice apply and additionally, applicants may seek the Agency’s input on topics relating specifically to orphan development such as:
The fees for scientific advice can be significant and details can be found on the EMA website. Significant fee reductions (up to 100%) are available for micro, small and medium size enterprises (SMEs) and for protocol assistance. DLRC group can help developers assess whether they may qualify for these fee reductions and are able to apply for SME status for the developer.
Scientific advice is co-ordinated and managed by the EMA’s Scientific Advice Working Party (SAWP) who delivers the advice after adoption at the regular Committee for Medicinal Products for Human Use (CHMP) meetings. There are approximately 11 opportunities per year to obtain scientific advice which is adopted at the monthly meetings of the CHMP. The timings are strict and applicants who miss the allocated submission dates (published on the EMA website) even if late by only one day, will need to wait until the next cycle.
There are programmes in place which allow parallel scientific advice from:
Further details can be found on the EMA Scientific Advice homepage.
When sponsors require advice relating to clinical trials, it is often sought via scientific advice from individual National Competent Authorities (NCAs) as NCAs are responsible for reviewing and approving clinical trial applications (CTAs) in the EU. More recently, as part of the initiative ‘Accelerating Clinical Trials in the EU (ACT-EU)’, two consolidated advice pilots are now available for developers requiring advice on clinical trials. The first is the SAWP-Clinical Trial Coordination Group (CTCG) pilot which provides advice on both clinical trials and marketing authorisation application requirements and the second is the Pre-CTA advice pilot. This pilot aims to provide technical and regulatory support on the dossier of a CTA prior to submission via the CT information system (CTIS).
An overview of the scientific advice procedure is given in the guidance document ‘European Medicines Agency Guidance for Applicants seeking scientific advice and protocol assistance (EMA/4260/2001 Rev. 14).’ The following figure summarises the two phases of the procedure.
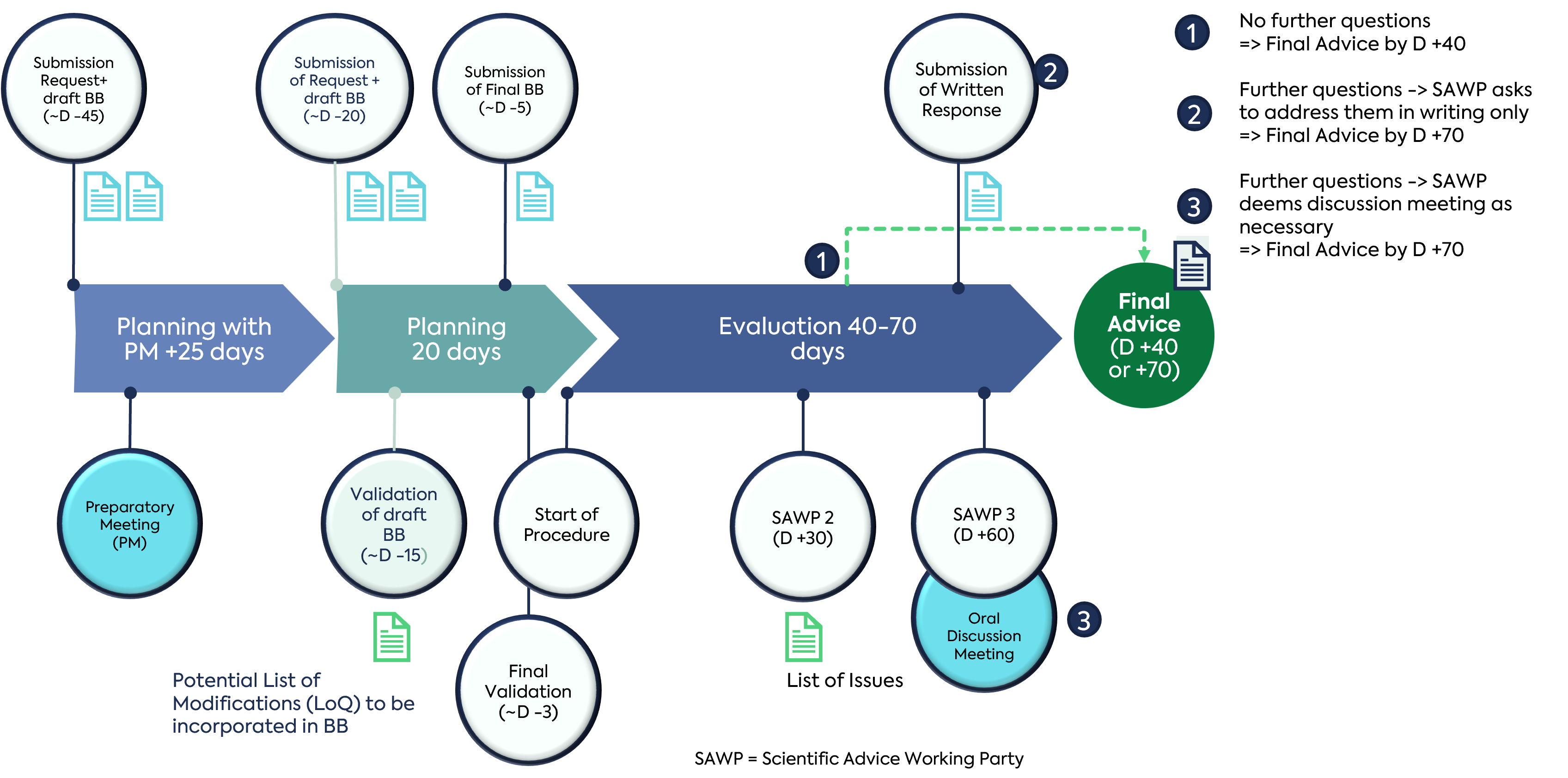
An optional preparatory telecon can be requested at the time of submission of the application for scientific advice. The preparatory meeting offers applicants the opportunity to establish contact with Agency staff and subject matter expert (SME) assessors and to receive feedback on several topics, including the appropriateness of the content and scope of questions and the related applicant’s positions, identification of additional issues to be included in the request, and the opportunity to ask regulatory questions which are outside the scope of scientific advice. This preparatory meeting may be the only direct communication that the applicant has with the agency and can be extremely useful. Many clients resist having a preparatory meeting due to the additional time required but this should be balanced against the benefit of a meeting which increases the likelihood of a Day 40 outcome, as the EMA has helped to shape the questions and briefing book content.
Our experience of preparatory meetings highlights the importance of succinct presentation that highlights the key points of the development and the rationale for the questions posed. We have supported clients in preparing for preparatory meetings in order to maximise their value. It is important to note that the preparatory meeting is about listening to feedback so that the briefing book and questions can be optimised and is NOT about defending the clients positions.
A unique feature of the procedure with the EMA is that the briefing book must be submitted with the initial application. Generally, for scientific advice with individual NCAs, the briefing book is submitted after the initial application and around 2-4 weeks before any advice meeting with the agency. There is a mandatory EMA template for the briefing book. The template provides guidance text, however our experience has helped clients identify the critical information to include, identifying the most appropriate questions and also develop a succinct stand-alone applicants position for each question.
To avoid unnecessary validation comments, it is important to consider carefully the wording of the proposed questions to be included in the advice request. Questions which are worded in the sense of a pre-assessment of data should be avoided.
The main purpose of this initial phase is to secure a briefing book which has been reviewed by the EMA and is found to be acceptable for the evaluation phase and is hence, validated.
After validation of the application, the evaluation process starts and is classified as Day 0. The reports are discussed at the next months SAWP meeting and then adopted at the next CHMP meeting which will be around Day 40. If the SAWP decides there is the need for a discussion meeting, this will be held after the CHMP meeting but before the next CHMP meeting, hence the advice will be adopted around Day 70 of the procedure.
If aspects of the written advice received are unclear or ambiguous, it is possible to request a clarification, with minor clarifications being addressed in writing in an expedited manner, and major clarifications being addressed at the following SAWP meeting. Any new data, information or a new applicant’s position would usually require a follow-up advice procedure.
The European Commission (EC) is proposing an ambitious revision of the EU pharmaceutical legislation which includes several points relating to Scientific Advice and proposes to optimise the regulatory support to SME and non-commercial organisations and to provide scientific advice for future MA applicants more generally and in greater depth. Structures allowing the development of advice for companies, in particular SMEs, should also be put in place.
Additional information on the impact of the Pharmaceutical Legislation Reform on Paediatric Development is available in our recent article.
There is a strong correlation between the success of marketing authorisation applications and applicants following EMA scientific advice, therefore seeking Scientific Advice during drug development is highly recommended. There are several routes to obtaining advice from Regulatory Authorities, such as advice from National Competent Authorities or EMA scientific advice (as summarised in this article.). In order to provide optimal value from seeking scientific advice, it is important for applicants to understand the nuances of the process, including the value of the preparatory meeting, the most appropriate questions to ask, how to provide optimal company positions in the briefing book, in addition to navigating the procedure and understanding if any follow-up actions are required.
If you are considering seeking scientific advice on your drug development program, DLRC’s extensive experience in helping clients navigate the scientific advice process will ensure that you obtain optimal input from the Regulators.
Contact DLRC’s team of award-winning regulatory professionals via the link below, or email hello@dlrcgroup.com.


Published Jul 21, 2025

Published Jul 11, 2025

Published Jul 07, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 01, 2025

Published Apr 28, 2025