DLRC Achieves Cyber Essentials Plus Certification
Published Apr 11, 2025
Published 01st August 2024

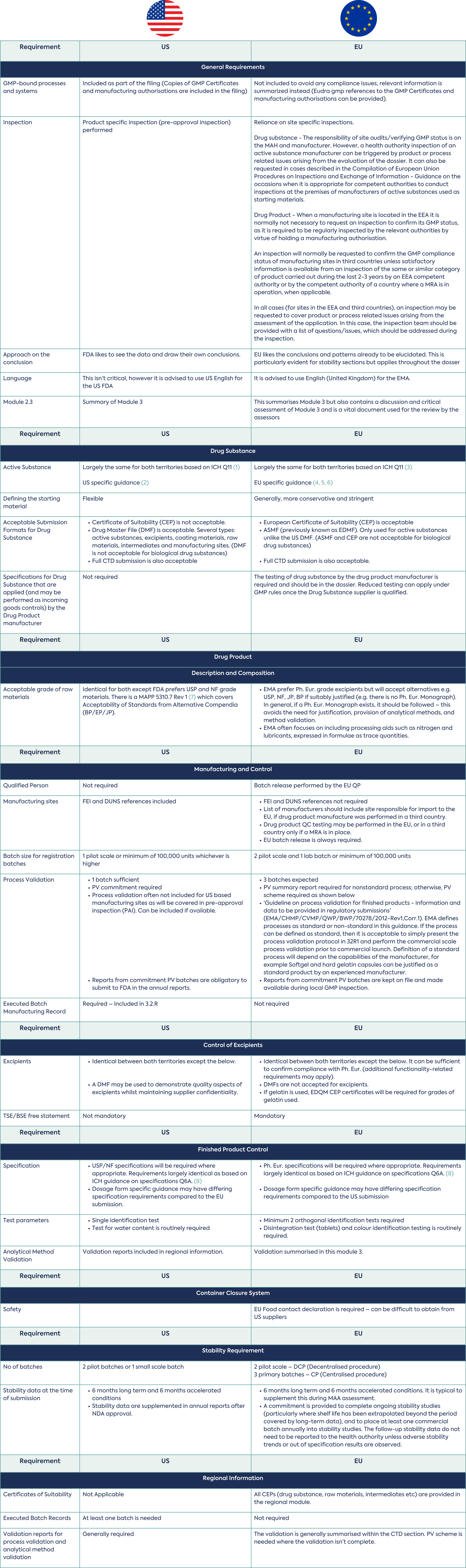
The European Union (EU) and the United States (US) are two of the most demanding regions in the world, often the priority for first registration, and are the starting points for the global dossier through optimised and harmonised requirements. The EU and the US follow different approval procedures which are unique in their own ways. Although they share a broadly common format and the common goal of patient safety, there are a few differences in the guidance, regulations and drug approval process that impact on the quality dossier and should be taken into account to assure successful registration of medicines in both regions.
The format for chemistry, manufacturing and controls (CMC) content of a marketing authorisation or new drug application dossier is presented for the EU in article 6 of EU Regulation (EC) No. 726/2004, and Annex I to Directive 2001/83/EC, legal requirements which are interpreted as ‘Notice to Applicants’, and for the US, in the FD&C Act and supporting regulations in Title 21 of the Code of Federal Regulations, incorporated as ‘Guidance for Industry’. These legal frameworks are supported by a series of nonbinding guidance documents discussed below, as well as mandatory quality standards described in the regional and national pharmacopoeias (Ph. Eur., USP and others) and requirements for good manufacturing practice (GMP).
Although the overarching requirements are similar in both regions, the interpretation and practical incorporation show significant differences which originate from the differing circumstances and mindsets at the respective authorities. This impacts on developers trying to meet the quality requirements in both regions.
Inter-regional co-operations were begun to harmonize international coordination, foremost being the International Conference on Harmonisation (ICH). Since its inception, the ICH has published many harmonised guidelines concerning all aspects of the quality, safety and efficacy of medicinal products. The detailed format of the CMC dossier is stipulated in ICH multidisciplinary guideline M4Q and the scientific content relating to medicinal products, their manufacturing, control and distribution is outlined in ICH guidelines Q1 to Q14. Since EU and US regions are members of ICH, they share this core body of scientific and regulatory guidelines. Additionally, GMP standards are well harmonised and for most product types and consequently the outcome of GMP inspections by US and EU authorities is recognised by each other via mutual recognition agreement (MRA).
Both of these health authorities continuously make efforts to harmonise their guidelines and compendial monographs’ content. In spite of this, there remain important differences in interpretation that have sometimes prevented new drugs from reaching patients in need.
The Food and Drug Administration (FDA) in the US and the European Medicines Agency (EMA) in Europe are the health authorities that regulate medicinal products. In the EU all regulations need to be coordinated between each member state.
This article will focus on the part describing CMC aspects of marketing authorisation applications which will examine the differences between these two regions.
The overview of the differences between the US and EU’s regulatory expectations is presented below:
Although the US and the EU are both ICH members and thus share many scientific and regulatory guidelines, there are significant differences in how these are interpreted and implemented.
In summary, it can be concluded that the identical legal framework (CTD) and regulatory guidelines on content and structure (ICH:M4) produce subtle but important different quality dossiers in the US and EU because of the differences in the mindset and expectations at the relevant health authorities.
DLRC has a wealth of experience in this area and is ready and willing to provide guidance on the information to be presented in the dossier for the US and EU regions. Our team of CMC experts can advise on regulations concerning EU and the US guidance. Please contact us using the links below for further information.


Published Apr 11, 2025

Published Mar 27, 2025

Published Mar 06, 2025
Published Feb 26, 2025

Published Feb 25, 2025

Published Feb 03, 2025

Published Feb 03, 2025

Published Jan 30, 2025

Published Jan 30, 2025